Estudo analisa dispersão do vírus da hepatite B nas Américas
 Ao menos 257 milhões de portadores crônicos da hepatite B em todo o mundo, segundo estimativas da Organização Mundial da Saúde. Pesquisadores do Instituto Oswaldo Cruz (IOC/Fiocruz) traçaram a evolução e dispersão de um dos perfis genéticos do vírus da hepatite B nas Américas, responsável por casos de desenvolvimento de câncer de fígado. “Amplamente distribuído no planeta, o vírus da hepatite B é dividido em pelo menos dez genótipos, que são identificados por letras de A a J. Com distribuição geográfica distinta, esses perfis genéticos apresentam diferenças significativas na progressão da doença, resposta à terapia antiviral e desfechos clínicos”, explicou Natalia Motta de Araujo, pesquisadora do Laboratório de Virologia Molecular do IOC e coordenadora do estudo.
Ao menos 257 milhões de portadores crônicos da hepatite B em todo o mundo, segundo estimativas da Organização Mundial da Saúde. Pesquisadores do Instituto Oswaldo Cruz (IOC/Fiocruz) traçaram a evolução e dispersão de um dos perfis genéticos do vírus da hepatite B nas Américas, responsável por casos de desenvolvimento de câncer de fígado. “Amplamente distribuído no planeta, o vírus da hepatite B é dividido em pelo menos dez genótipos, que são identificados por letras de A a J. Com distribuição geográfica distinta, esses perfis genéticos apresentam diferenças significativas na progressão da doença, resposta à terapia antiviral e desfechos clínicos”, explicou Natalia Motta de Araujo, pesquisadora do Laboratório de Virologia Molecular do IOC e coordenadora do estudo.
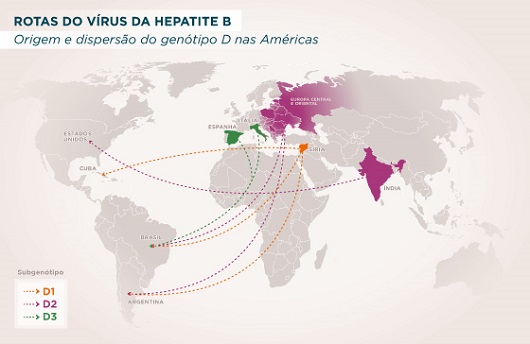
Conhecido como genótipo D, este perfil também está relacionado a baixas respostas ao tratamento pelo interferon, um dos principais medicamentos usados no combate à infecção. Após análise de dados do sequenciamento completo do genoma do vírus, realizado pelo grupo a partir de amostras brasileiras, e de sequências genéticas de países do continente americano disponíveis em um banco de dados internacional, os cientistas apontam que as principais rotas de introdução do genótipo D na região estão relacionadas a movimentos de migração em massa, entre os séculos 19 e 20, com origem principalmente na Síria, Líbano, Índia e em países da Europa Central e Oriental.
“Conhecer as atividades humanas relacionadas à dispersão de um genótipo é importante para a vigilância em saúde, com definições, por exemplo, de estratégias de prevenção. Ampliar o conhecimento dos genótipos que circulam numa região contribui para o desenvolvimento de estudos acerca de diferenças clínicas e de respostas terapêuticas que podem ocorrer devido à genética do vírus”, destacou a pesquisadora. A introdução do genótipo D do vírus da hepatite B nas Américas tem como origem principal a Síria, Líbano, Índia, Espanha, Itália e países da Europa Central e Oriental.
Panorama genético
Os pesquisadores realizaram o sequenciamento completo do genoma de 39 amostras, das cinco regiões do Brasil, de pacientes infectados com o genótipo D do vírus da hepatite B. Somaram-se à análise 1.135 sequências (209 completas e 926 parciais) isoladas no Brasil, Argentina, Canadá, Chile, Colômbia, Cuba, Haiti, Martinica, México, Estados Unidos e Venezuela. Com base em análises filogenéticas – uma espécie de árvore genealógica do vírus -, os pesquisadores estabeleceram um panorama ainda mais detalhado considerando os subgenótipos que circulam no Brasil e nas Américas. Eles descobriram que, dos dez subgenótipos (do genótipo D) existentes, pelo menos cinco circulam no continente americano (D1, D2, D3, D4 e D7). Destes, apenas o D7 não foi identificado como circulante no Brasil. O subgenótipo D1 foi o mais prevalente na Argentina e Canadá, assim como o D2 nos Estados Unidos, o D3 no Brasil, e o D4 em Cuba e no Haiti.
O que diferencia esses subgenótipos são características bem específicas. Para estimar a origem e data de introdução dos [subgenótipos] circulantes nas Américas, os pesquisadores analisaram as sequências genéticas com o uso de ferramentas de bioinformática que consideram principalmente o local de origem e a data de coleta das amostras. A reconstrução espaço-temporal mostrou que o D1 possivelmente tenha origem na Síria, de onde chegou ao Brasil, Cuba e Argentina em ocasiões diferentes. Já a análise filogeográfica do D2 sugeriu que o mais provável epicentro tenha sido a Europa Central e Oriental, com destaque para a Rússia, incluindo países como Estônia, Polônia e Sérvia. As sequências genéticas dos Estados Unidos, quando agrupadas com as indianas, apontaram que pelo menos duas introduções diferentes deste subgenótipo ocorreram no país norte-americano a partir da Índia. O principal período de introdução foi estimado entre 1966 e 1978.
O subgenótipo D3 apresentou vias de dispersão complexas, incluindo diferentes regiões geográficas e múltiplas introduções. As análises destacaram que o D3 tenha como origem provável o Sul da Europa (Itália e Espanha) e o Brasil como fonte de dispersão para o continente. O subgenótipo D3 foi encontrado em todas as regiões do Brasil, com as mais altas taxas na Região Sul, que recebeu um fluxo intenso de imigrantes europeus. A investigação do D4 demonstrou uma maior prevalência desse subgenótipo em países americanos onde a população majoritária é de descendentes de africanos. Entretanto, os resultados sobre o local de origem do D4 foram inconclusivos. “Somente não foi possível desenhar a árvore filogenética do subgenótipo D7, devido à sua natureza recombinante e ao número limitado de sequências disponíveis”, ponderou a especialista.
Os pesquisadores destacaram que, de forma majoritária, os achados foram compatíveis com dados históricos e epidemiológicos, demonstrando assim a utilidade das ferramentas de bioinformática em estudos evolutivos. A migração europeia em massa para as Américas ocorreu do início do século 19 até meados do século 20, especialmente para os Estados Unidos, Argentina, Canadá, Brasil, Cuba e Uruguai. Em período semelhante, milhares de pessoas, principalmente da Síria, Líbano, Palestina, Turquia e Egito migraram para Cuba. Em relação ao Brasil, o movimento migratório de árabes, em especial de países como Síria e Líbano, também é documentado a partir da segunda metade do século 19. Já nos Estados Unidos, um dos maiores grupos de imigrantes tem como origem a Índia.
O estudo foi realizado como parte do projeto de doutorado da estudante Natália Spitz Toledo Dias, do Programa de Pós-graduação Stricto sensu em Biologia Parasitária do IOC. “As análises também permitiram conhecer melhor a velocidade com que o vírus evolui e a forma como ele vai se modificando ao longo do tempo”, frisou. “Determinadas mutações são importantes do ponto de vista clínico, uma vez que podem gerar resistência a um medicamento antiviral, falha na resposta à vacina ou fazer com que o microrganismo não seja reconhecido nos kits de diagnóstico, por exemplo”, completou.
A pesquisa, publicada no periódico científico Plos One (em inglês), também contou com a contribuição dos pesquisadores do IOC Gonzalo Bello, do Laboratório de Aids e Imunologia Molecular; Selma Gomes, do Laboratório de Virologia Molecular; Francisco Mello, do Laboratório de Hepatites Virais, e Aline Moreira, do Laboratório de Genômica Funcional e Bioinformática. Participaram, ainda, especialistas da Universidade Federal de Goiás (UFG) e da Fundação Estadual de Produção e Pesquisa em Saúde (FEPPS), do Rio Grande do Sul.